ggheatmapper: Tile-able heatmaps that play well with ggplots
Clarice S. Groeneveld
2026-01-27
ggheatmapper.RmdIntroduction
ggheatmapper is built using ggplot2 and the
patchwork framework for aligning plots. It supports adding
into the heatmap and all the powerful tweaks enabled by
themes in ggplot, and helps you align more information
about your data to the original heatmap to make “complex heatmaps” with
a more modern and tweakable framework.
Using a matrix as your table
The simplest, yet less powerful, way to use ggheatmapper
is with a matrix as-is. Here, we demonstrate with a subset of a gene
expression matrix for bladder cancer samples:
library(tidyverse)
#> ── Attaching core tidyverse packages ──────────────────────── tidyverse 2.0.0 ──
#> ✔ dplyr 1.1.4 ✔ readr 2.1.6
#> ✔ forcats 1.0.1 ✔ stringr 1.6.0
#> ✔ ggplot2 4.0.1 ✔ tibble 3.3.0
#> ✔ lubridate 1.9.4 ✔ tidyr 1.3.2
#> ✔ purrr 1.2.0
#> ── Conflicts ────────────────────────────────────────── tidyverse_conflicts() ──
#> ✖ dplyr::filter() masks stats::filter()
#> ✖ dplyr::lag() masks stats::lag()
#> ℹ Use the conflicted package (<http://conflicted.r-lib.org/>) to force all conflicts to become errors
library(patchwork)
library(ggheatmapper)
data(tcgaBLCA_ex)
gexp <- tcgaBLCA_ex$gexp
gghm <- ggheatmap(gexp,
hm_colors = 'RdBu',
hm_color_values = scales::rescale(c(-4,-2,-1,-0.5,-0.25,0,0.25,0.5,1,2,4,6)),
scale = TRUE,
center = TRUE,
show_dend_row = TRUE,
colors_title = "Scaled expression (log2 UQ)",
show_colnames = FALSE)
#> Running `ggheatmap` in matrix mode. If that's not intentional, provide a `colv`.
gghm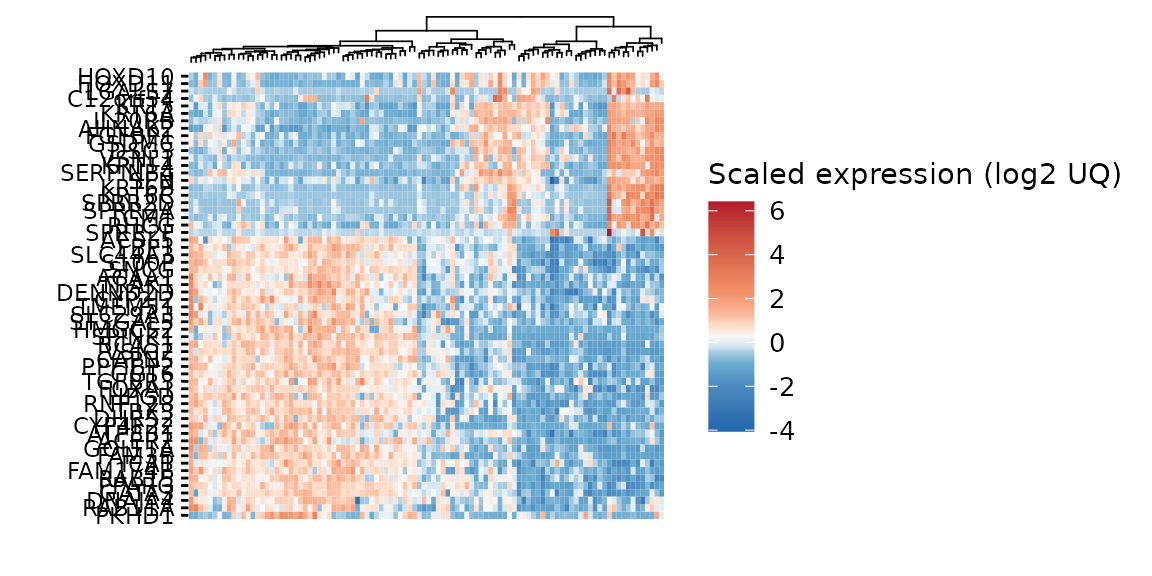
This method still will support extending with
align_to_hm but would not support add_tracks.
You can still modify this by using & and adding
theme, for example:
gghm &
theme(axis.text = element_text(size = 6))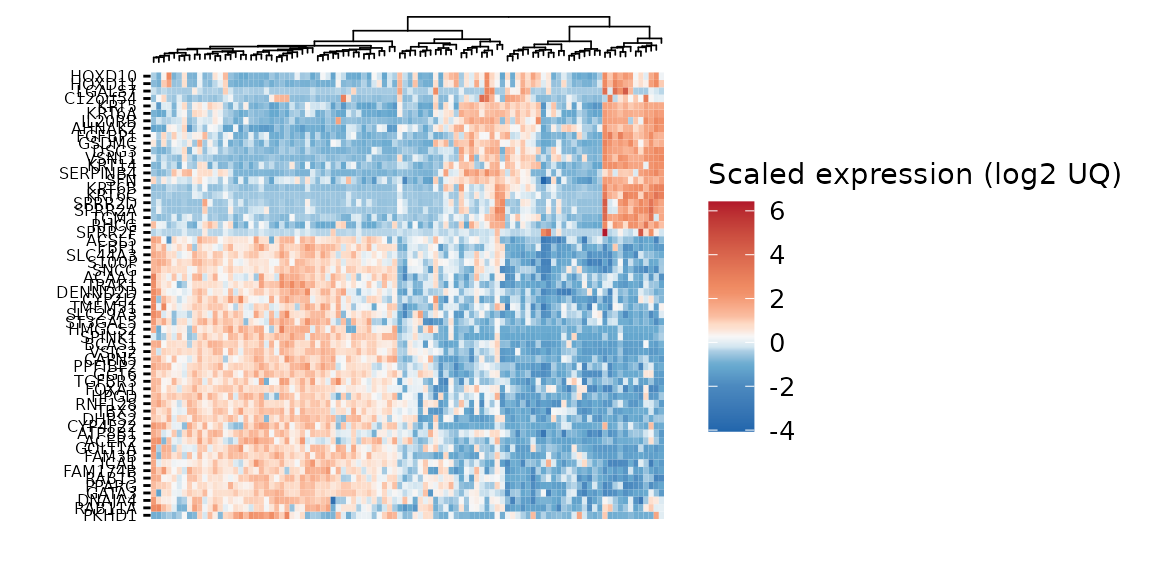
Using tables with more variables
If transpose our original table and extended it with new variables,
we can unlock more features using ggheatmap.
sample_annot <- tcgaBLCA_ex$sample_annot %>% as_tibble()
genes <- rownames(gexp)
tcgaBLCA_tb <- gexp %>%
t() %>%
as.data.frame() %>%
rownames_to_column("sample") %>%
left_join(sample_annot, by = "sample") %>%
tibble() %>%
group_by(consensusClass)
head(tcgaBLCA_tb)| sample | FBP1 | ACER2 | PKHD1 | CAPN5 | S100P | TMEM51 | DHRS2 | CYP4F22 | SPINK1 | ACSL5 | ST3GAL5 | TBX3 | HPGD | TGFBR3 | FAM3B | ATP8B1 | RNF128 | SNCG | SLC44A3 | GATA3 | PPARG | ICA1 | GGT6 | RAB11A | TRAK1 | VSIG2 | BCAS1 | RAB15 | FAM174B | SLC29A3 | FOXA1 | GOLT1A | PPFIBP2 | DENND2D | ACAA1 | DNAJA4 | HMGCS2 | CYP2J2 | VSNL1 | KRT14 | TGM1 | SERPINB4 | GSDMC | KRT6A | LGALS7 | SFN | SPRR2A | C12orf54 | SPRR2D | HOXD11 | KRT6C | KRT5 | DSG3 | KRT6B | HOXD10 | IL20RB | RHCG | AHNAK2 | SPRR2F | FGFBP1 | sex | age | stage | node | metastasis | papillary | squamous | neuroendocrine | plasmacytoid | consensusClass | cor_pval | separationLevel | LumP | LumNS | LumU | Stroma.rich | Ba.Sq | NE.like |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| TCGA-XF-A9SW-01A | 3.051428 | 0.7600492 | 1.0344884 | 3.190462 | 7.252360 | 4.695958 | 3.553598 | 0.0458037 | 8.070943 | 5.222600 | 2.7737241 | 4.846704 | 4.195551 | 5.317267 | 3.1750867 | 4.278225 | 3.175087 | 4.8426545 | 2.011588 | 5.840220 | 4.209453 | 2.770317 | 3.728798 | 5.853159 | 4.839407 | 3.2770249 | 0.9406215 | 4.783051 | 3.517479 | 3.205675 | 2.9853829 | 3.279423 | 2.467868 | 3.198088 | 3.772022 | 3.682428 | 0.3495844 | 0.718229 | 0.6896599 | 0.8271634 | 0.5215371 | 0.0230836 | 1.3219281 | 0.2937312 | 0.0000000 | 6.262246 | 1.2842084 | 0.3856537 | 0.2552571 | 0.1118929 | 0.0230836 | 1.853159 | 0.0000000 | 0.0000000 | 0.3677318 | 0.5052353 | 1.8786937 | 5.6623525 | 0.0000000 | 0.0901978 | M | 85 | T3 | N2 | NA | 0 | 0 | 0 | 0 | Stroma-rich | 0 | 0.8871657 | 0.3931646 | 0.4925222 | 0.4609638 | 0.6421217 | 0.4954034 | 0.2989414 |
| TCGA-BL-A13J-01B | 2.538652 | 4.4439186 | 0.2504068 | 2.982211 | 4.283709 | 2.692439 | 8.245226 | 1.7312968 | 3.901222 | 4.771899 | 3.2720427 | 5.745076 | 5.665011 | 4.000000 | 1.6374299 | 5.595922 | 5.496664 | 1.4043903 | 3.260282 | 5.052941 | 5.196140 | 3.543512 | 2.441317 | 5.449971 | 5.443269 | 3.0979633 | 1.8087013 | 3.592799 | 3.937369 | 1.390071 | 2.4585741 | 1.371969 | 3.780845 | 3.206136 | 3.224412 | 6.407171 | 2.1814040 | 2.950846 | 0.0554951 | 3.5974805 | 0.9714308 | 0.4360991 | 0.6614754 | 2.7732793 | 0.0000000 | 3.995869 | 1.0093987 | 0.0187366 | 0.7026141 | 0.5499671 | 0.0372329 | 4.684393 | 1.3756074 | 0.2264279 | 1.1477536 | 0.9952776 | 2.3462385 | 4.5877101 | 0.0093987 | 0.6909794 | M | 65 | T4 | N2 | M0 | 1 | 0 | 0 | 0 | LumP | 0 | 0.8960881 | 0.5418487 | 0.4977683 | 0.5031495 | 0.4995555 | 0.4950211 | 0.1822938 |
| TCGA-C4-A0F1-01A | 1.532221 | 2.0437214 | 3.0887679 | 2.473487 | 4.091374 | 3.335184 | 4.318595 | 0.2255597 | 1.804181 | 1.606989 | 0.8845228 | 2.225560 | 1.054448 | 1.599684 | 0.1475572 | 4.381709 | 3.059781 | 0.9888594 | 2.791413 | 4.214842 | 1.739183 | 2.461448 | 1.022026 | 6.647846 | 5.364572 | 0.1273793 | 0.2255597 | 1.562595 | 1.524527 | 2.191951 | 0.2630344 | 0.000000 | 2.592342 | 3.752419 | 4.852614 | 5.689729 | 0.0000000 | 2.566347 | 5.9933957 | 14.1342548 | 1.5700892 | 8.6758485 | 4.1235643 | 13.8688908 | 0.3526716 | 10.493024 | 4.8144715 | 0.5923420 | 5.0302002 | 3.5341382 | 10.4124884 | 13.898507 | 10.7761166 | 11.8531476 | 4.1056265 | 8.1470875 | 6.8639003 | 10.1725237 | 0.4533656 | 6.4604401 | M | 71 | T3 | N0 | M0 | 0 | 1 | 0 | 0 | Ba/Sq | 0 | 0.8163011 | 0.2997577 | 0.2315946 | 0.2387890 | 0.3410051 | 0.6597589 | 0.1615142 |
| TCGA-BT-A20W-01A | 5.978460 | 5.0488941 | 1.2202661 | 4.915787 | 7.032628 | 4.972314 | 8.033026 | 5.0979235 | 8.851008 | 6.831062 | 5.1470213 | 7.443114 | 6.536437 | 4.238503 | 4.1879897 | 5.445847 | 5.888681 | 8.1717314 | 4.009266 | 7.773360 | 6.569229 | 4.644451 | 6.227231 | 6.769685 | 6.237912 | 5.8543865 | 5.1486993 | 5.892191 | 3.999070 | 4.605880 | 5.4792386 | 2.973734 | 5.580307 | 4.923159 | 5.437634 | 5.724268 | 6.6118234 | 2.794550 | 0.1280076 | 2.5370783 | 0.9314686 | 0.0000000 | 0.2949049 | 0.9157870 | 0.0000000 | 5.650089 | 0.0294438 | 0.1143327 | 0.1143327 | 0.5599585 | 0.0147970 | 2.134797 | 0.0725125 | 0.0865877 | 0.8009666 | 0.7399372 | 0.4335102 | 1.3009540 | 0.0000000 | 0.1814469 | M | 71 | T2 | N0 | M0 | 0 | 0 | 0 | 0 | LumU | 0 | 0.0257908 | 0.7403699 | 0.7359217 | 0.7420607 | 0.6170841 | 0.4691149 | 0.2982330 |
| TCGA-GV-A40G-01A | 6.027991 | 3.9534802 | 2.8433655 | 7.060945 | 6.679805 | 5.813682 | 4.730013 | 5.1503722 | 9.804690 | 3.986061 | 3.8487674 | 7.189749 | 6.635844 | 4.667193 | 4.3252536 | 5.299278 | 6.732415 | 8.4981895 | 3.684128 | 5.840269 | 5.940347 | 4.537487 | 5.706403 | 7.319151 | 6.190736 | 5.8231222 | 5.0257784 | 6.155207 | 4.876234 | 4.512908 | 4.8017954 | 2.762267 | 5.332157 | 4.636290 | 5.964363 | 5.688439 | 5.1330829 | 2.457413 | 0.1171835 | 2.3351842 | 1.6322682 | 0.3699496 | 0.2444187 | 0.2160369 | 0.0110552 | 6.640857 | 0.1674567 | 0.1475572 | 0.0756643 | 0.7458165 | 0.0220263 | 2.471488 | 0.0000000 | 0.0756643 | 1.1968007 | 0.6849913 | 1.3996970 | 0.1968007 | 0.0000000 | 0.7785321 | M | 77 | T2 | N0 | NA | 0 | 0 | 0 | 0 | LumU | 0 | 0.0748451 | 0.6734381 | 0.6157098 | 0.6832107 | 0.4895690 | 0.4171794 | 0.2647129 |
| TCGA-HQ-A2OE-01A | 5.562190 | 5.3558523 | 3.5948780 | 5.413716 | 7.054534 | 5.541404 | 6.580083 | 1.1238717 | 7.464341 | 6.386977 | 5.2531547 | 9.160480 | 8.112341 | 5.058496 | 6.3492774 | 7.093522 | 5.697183 | 7.7692337 | 4.639871 | 7.434341 | 6.044997 | 3.523129 | 4.547161 | 7.047030 | 6.513904 | 4.4078684 | 5.1383513 | 6.474951 | 4.267174 | 4.653376 | 7.2894147 | 5.716423 | 6.040620 | 5.575813 | 5.173789 | 5.508778 | 9.2317177 | 7.126793 | 0.0099155 | 0.6842771 | 0.7449034 | 0.0774788 | 0.3436529 | 0.5682838 | 0.0000000 | 8.060994 | 0.0962153 | 0.2037952 | 0.9338331 | 0.0099155 | 0.0099155 | 4.418143 | 0.6529809 | 0.1599409 | 0.2630344 | 0.5548005 | 1.1820347 | 0.3975197 | 0.0000000 | 0.1329739 | M | 69 | T2 | N2 | NA | 1 | 0 | 0 | 0 | LumP | 0 | 0.3120613 | 0.6270255 | 0.5555841 | 0.5852272 | 0.4305816 | 0.4076061 | 0.2394613 |
One of the additional features we unlock is using
grouping with group_by to make semi-supervised
heatmaps. In this case, we need to say which column contains the IDs
that will be the columns of the heatmap (in this case,
colv = 'sample'), and what are the columns we want to plot
as rows (rowv = genes). Other parameters here are
graphical, for demonstration:
gr_gghm <- ggheatmap(tcgaBLCA_tb,
colv = "sample",
rowv = genes,
hm_colors = 'RdBu',
hm_color_values = scales::rescale(c(-4,-2,-1,-0.5,-0.25,0,0.25,0.5,1,2,4,6)),
scale = TRUE,
center = TRUE,
show_dend_row = FALSE,
show_colnames = FALSE,
show_rownames = FALSE,
group_colors = c(`Ba/Sq` = "#fe4a49", LumNS = "#32837d", LumP = "#06d6a0", LumU = "#009fb7",
`Stroma-rich` = "#f9c80e", `NE-like` = "#7d5ba6"),
colors_title = "Scaled expression (log2 UQ)")
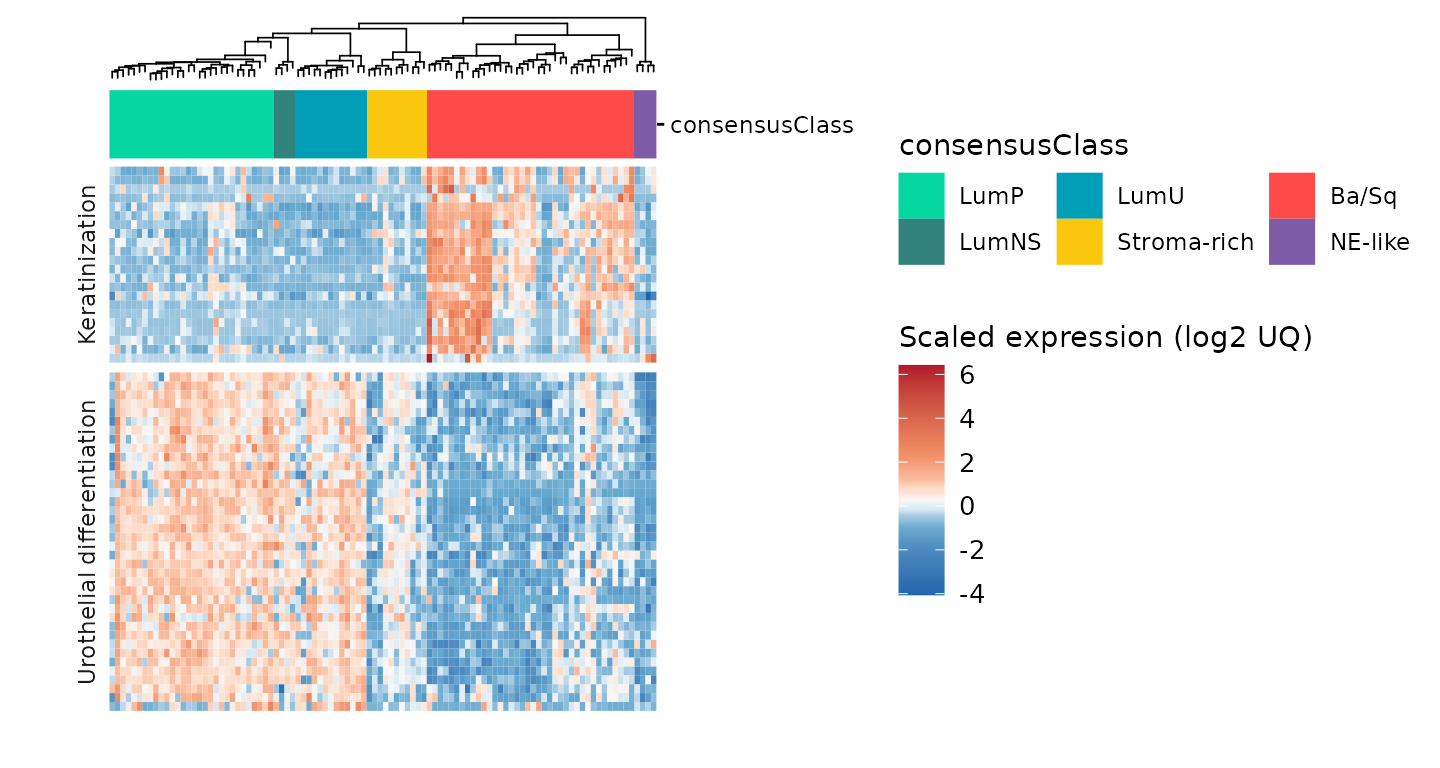
gr_gghm +
plot_layout(guides = 'collect')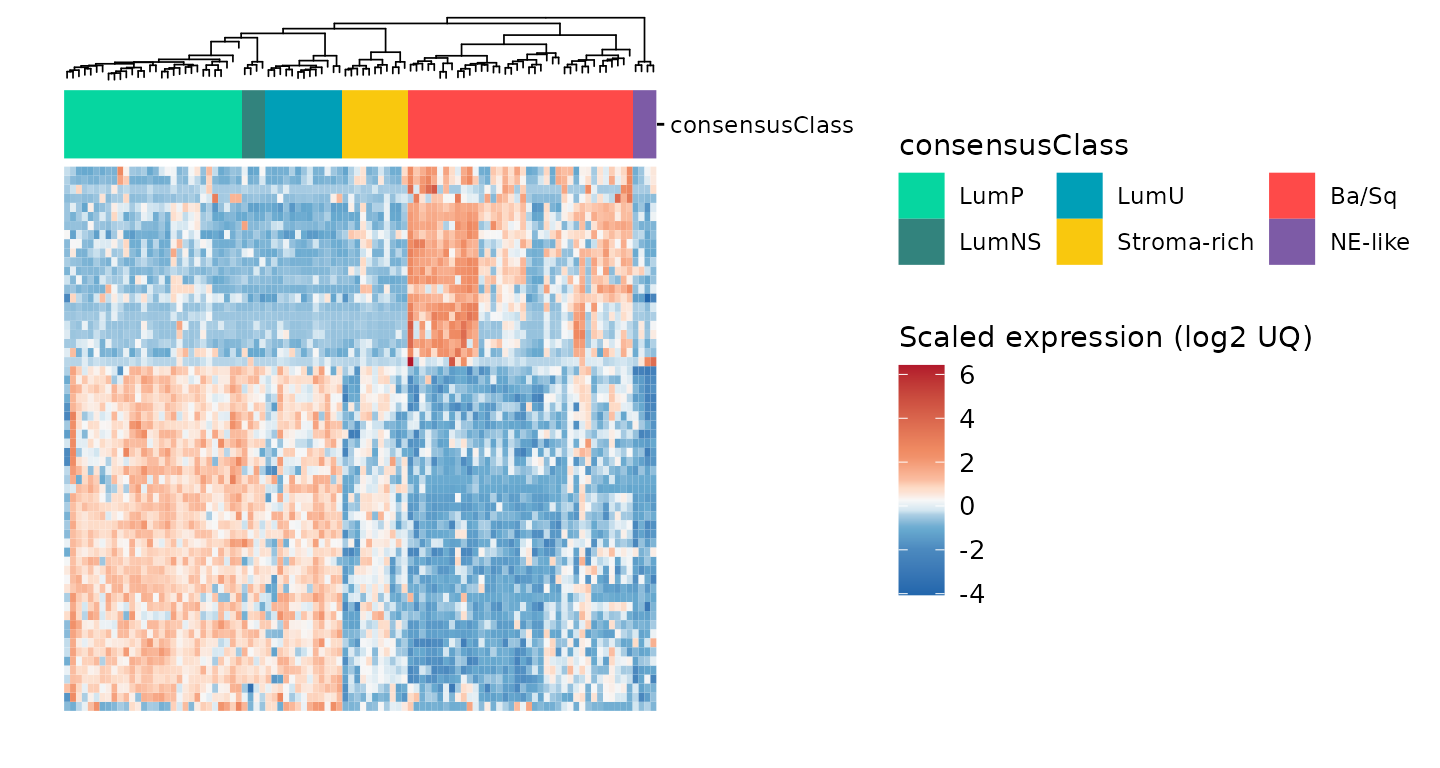
Here, we can see that the heatmap is clustered in a semi-supervised
manner (by consensusClass). It also enables
add_tracks:
Adding tracks
You can add_tracks for variables that were in the
original table fed to ggheatmap. You can see what variables
are available using get_data:
get_data(gr_gghm) %>% colnames()
#> [1] "observations" "FBP1" "ACER2" "PKHD1"
#> [5] "CAPN5" "S100P" "TMEM51" "DHRS2"
#> [9] "CYP4F22" "SPINK1" "ACSL5" "ST3GAL5"
#> [13] "TBX3" "HPGD" "TGFBR3" "FAM3B"
#> [17] "ATP8B1" "RNF128" "SNCG" "SLC44A3"
#> [21] "GATA3" "PPARG" "ICA1" "GGT6"
#> [25] "RAB11A" "TRAK1" "VSIG2" "BCAS1"
#> [29] "RAB15" "FAM174B" "SLC29A3" "FOXA1"
#> [33] "GOLT1A" "PPFIBP2" "DENND2D" "ACAA1"
#> [37] "DNAJA4" "HMGCS2" "CYP2J2" "VSNL1"
#> [41] "KRT14" "TGM1" "SERPINB4" "GSDMC"
#> [45] "KRT6A" "LGALS7" "SFN" "SPRR2A"
#> [49] "C12orf54" "SPRR2D" "HOXD11" "KRT6C"
#> [53] "KRT5" "DSG3" "KRT6B" "HOXD10"
#> [57] "IL20RB" "RHCG" "AHNAK2" "SPRR2F"
#> [61] "FGFBP1" "sex" "age" "stage"
#> [65] "node" "metastasis" "papillary" "squamous"
#> [69] "neuroendocrine" "plasmacytoid" "consensusClass" "cor_pval"
#> [73] "separationLevel" "LumP" "LumNS" "LumU"
#> [77] "Stroma.rich" "Ba.Sq" "NE.like"observations will always be your ID variable, which is a
factor ordered in the same way as the heatmap, to ease making new plots
that will be perfectly aligned. Here, we’ll add some clinical
tracks:
gr_gghm <- add_tracks(gr_gghm,
track_columns = c("stage", "node", "metastasis"),
track_colors = list(stage = 'Greys', node = 'Oranges', metastasis = 'Reds'),
track_prop = 0.2)
#> Adding missing grouping variables: `consensusClass`
gr_gghm +
plot_layout(guides = 'collect')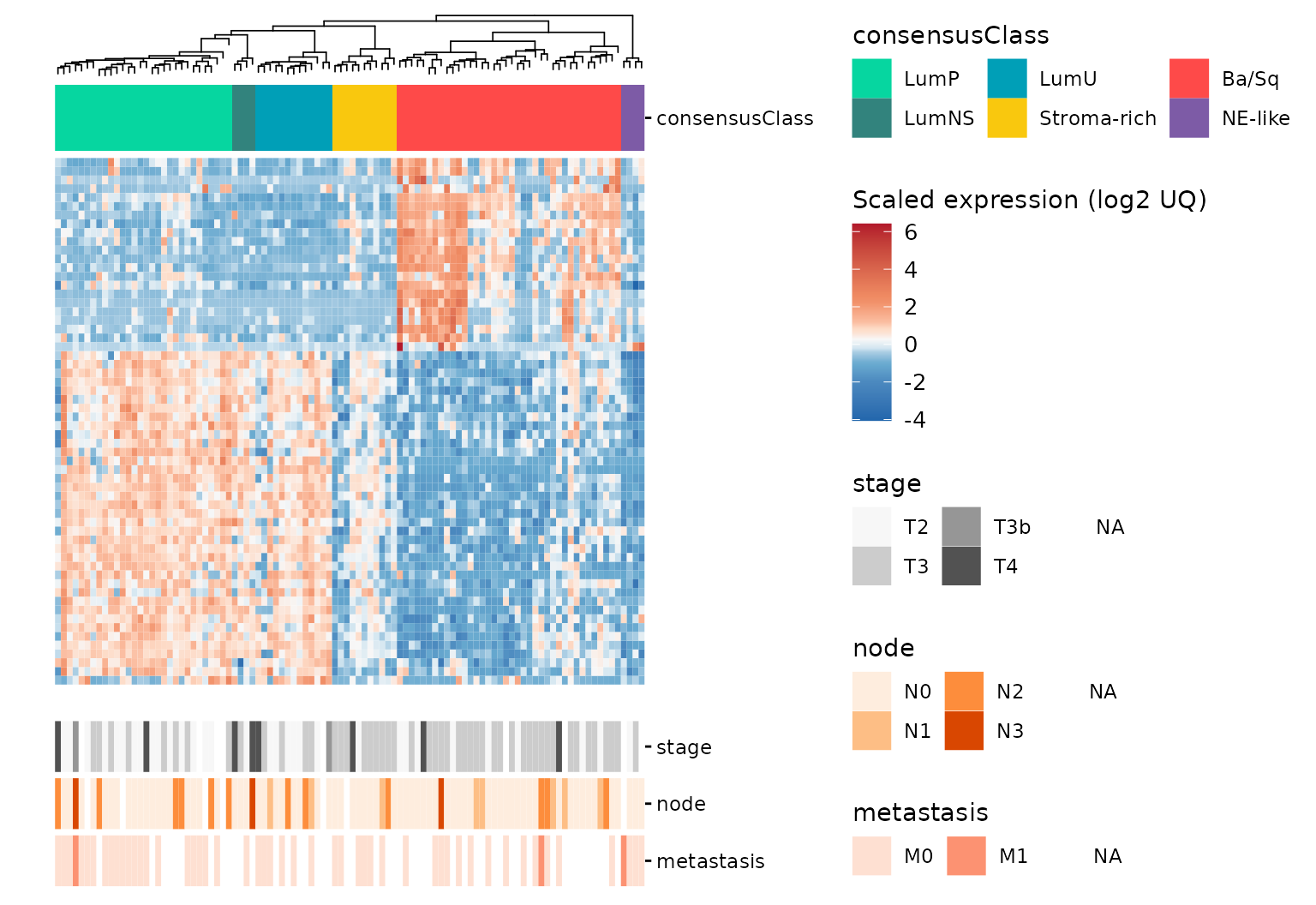
Aligning new plots
Here we’ll make two different plots: one to align with the samples
(columns) and another to align with the genes (rows). We can get the
data from get_data, which is a good idea to facilitate further plotting
because it will ensure your observations will be in the correct order.
Then, here, we make a line-plot with the correlations of each sample to
the centroid of the 6 consensus bladder cancer subtypes. Note the use of
theme_quant, one of our suggested themes that look nice
with ggheatmaps, and that we switch the y-axis to the right
side to make the end-product look better (though everything will work
without this):
tcgaBLCA_tb2 <- get_data(gr_gghm)
plt_corlines <- tcgaBLCA_tb2 %>%
ungroup() %>%
select(observations, LumP:NE.like) %>%
pivot_longer(cols = -observations, names_to = "subtype", values_to = "cor") %>%
ggplot(aes(observations, cor, color = subtype, group = subtype)) +
geom_line() +
scale_y_continuous(position = "right") +
scale_color_manual(values = c(`Ba.Sq` = "#fe4a49", LumNS = "#32837d", LumP = "#06d6a0", LumU = "#009fb7",
`Stroma.rich` = "#f9c80e", `NE.like` = "#7d5ba6")) +
guides(color = FALSE) +
labs(y = "Correlation\n to centroid") +
theme_quant()
#> Warning: The `<scale>` argument of `guides()` cannot be `FALSE`. Use "none" instead as
#> of ggplot2 3.3.4.
#> This warning is displayed once per session.
#> Call `lifecycle::last_lifecycle_warnings()` to see where this warning was
#> generated.
plt_corlines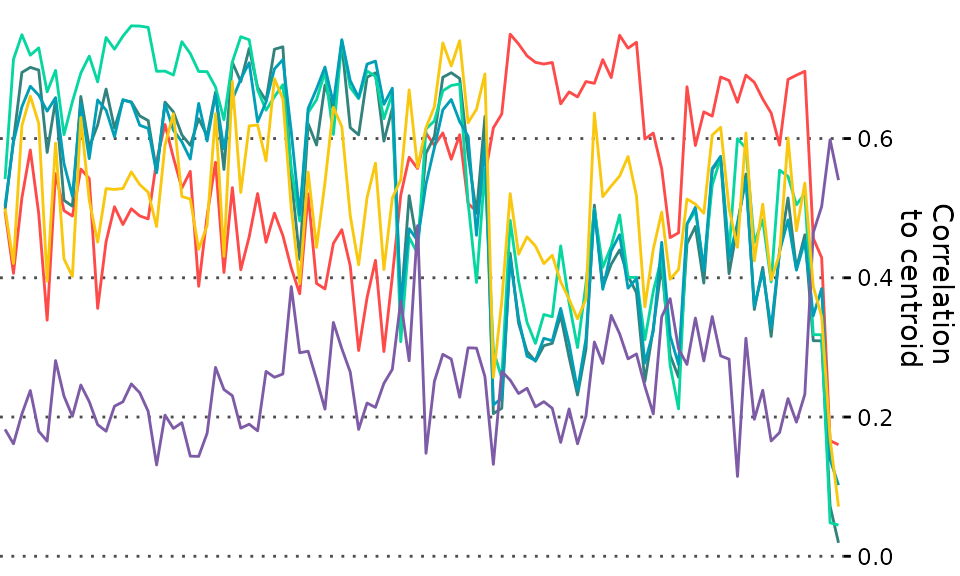
The second plot should align to the rows. We can’t just use the
get_data because it doesn’t contain the information to
order our rows, but we can get this from get_rowLevels.
Here, we plot which signature each gene in our example data belongs
to:
plt_row_annot <- tcgaBLCA_ex$gene_annot %>%
mutate(gene_symbol = factor(gene_symbol, levels = get_rowLevels(gr_gghm)),
group = 'signature') %>%
ggplot(aes(gene_symbol, group, fill = signature)) +
geom_tile() +
labs(y = "") +
coord_flip() +
theme_sparse2()
plt_row_annot
Finally, we can use align_to_hm to add these plots to
the original hm with all panels properly aligned. Note the use of
legend_action = 'collect' in the final call, that will
unite all the legends in a nice way:
gghm_complete <- gr_gghm %>%
align_to_hm(plt_corlines, newplt_size_prop = 0.3) %>%
align_to_hm(plt_row_annot, pos = "left", newplt_size_prop = 0.08,
legend_action = "collect", tag_level = 'keep')
gghm_complete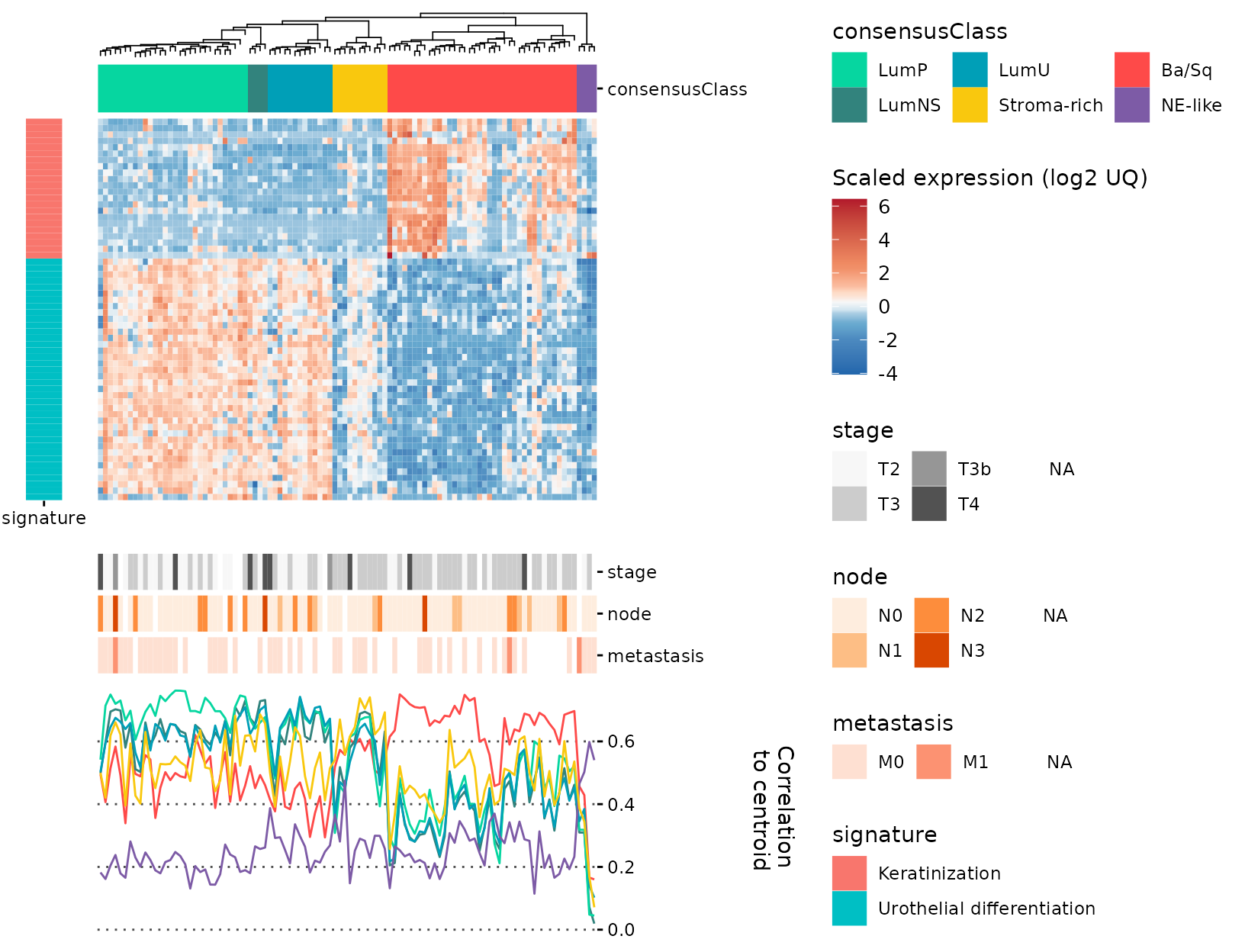
Note that you can still use the patchwork
& to make global changes to all plots:
gghm_complete <- gghm_complete &
theme(legend.text = element_text(size = 7),
legend.title = element_text(size = 8))
gghm_complete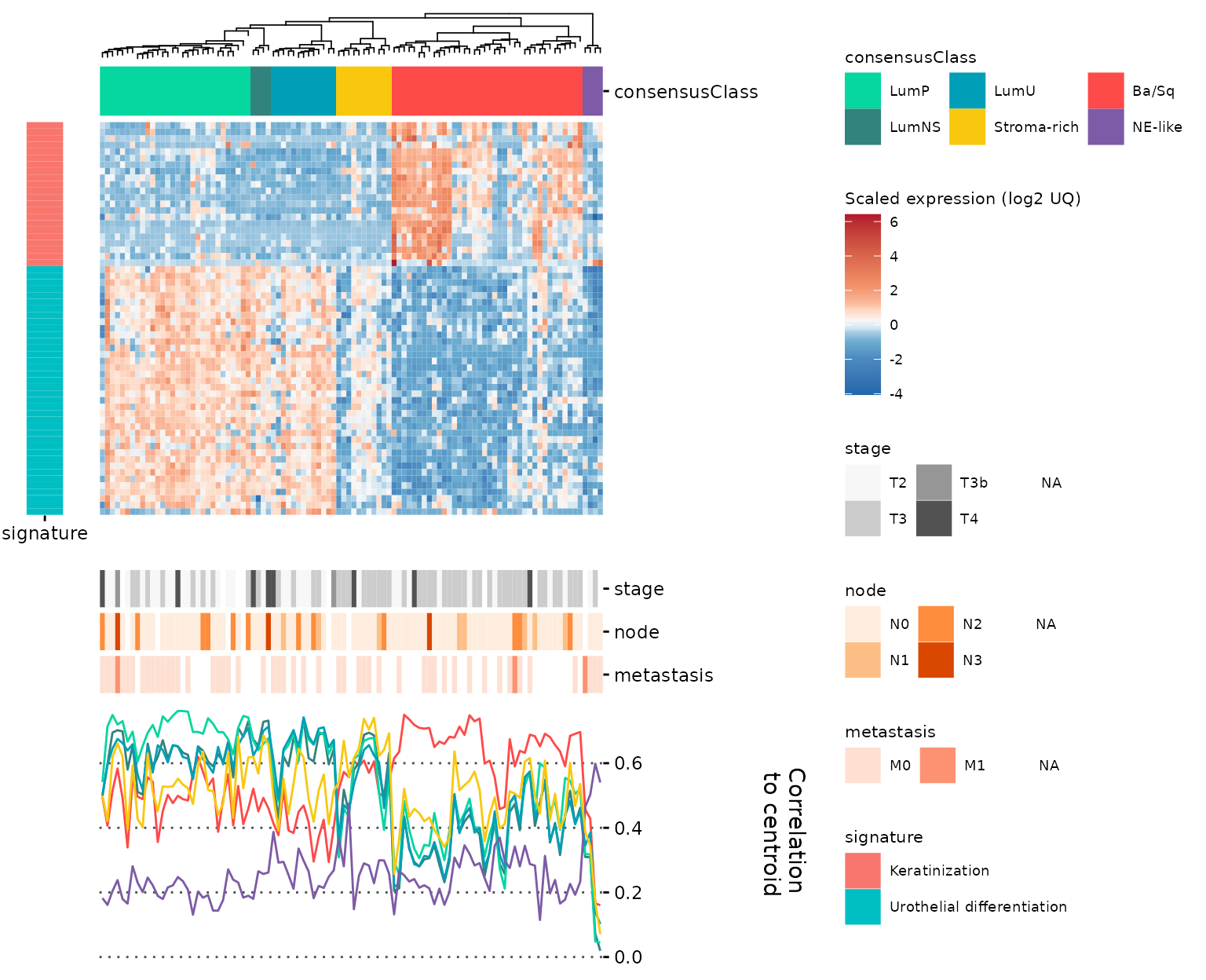
Creating panels
You can now make panels with your complete heatmap with
patchwork, cowplot or other aligning packages.
For example:
plt_subtype_count <- ggplot(sample_annot, aes(consensusClass, fill = consensusClass)) +
geom_bar() +
scale_fill_manual(values = c(`Ba/Sq` = "#fe4a49", LumNS = "#32837d",
LumP = "#06d6a0", LumU = "#009fb7",
`Stroma-rich` = "#f9c80e",
`NE-like` = "#7d5ba6")) +
labs(y = 'Number of samples') +
guides(fill = FALSE) +
theme_quant() +
theme(axis.ticks.x = element_line(color = "black"),
axis.text.x = element_text(color = "black", angle = 45, hjust = 1, vjust = 1))
plt_subtype_count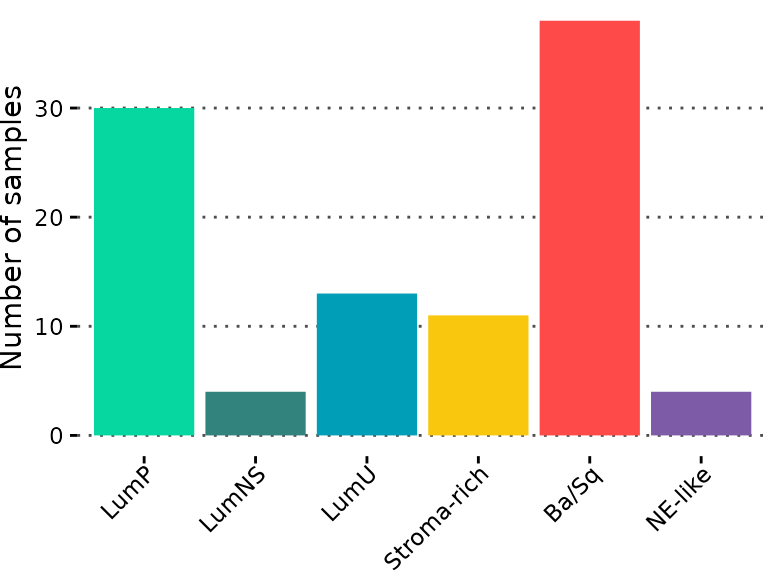
Then, we can just use standard patchwork to align the
ggheatmap and our new plots (as they won’t be aligned with
the heatmap part of the plot, but with the entire plot):
library(patchwork)
new_col <- (plt_subtype_count + plot_spacer()) +
plot_layout(heights = c(0.3,0.7))
(new_col | gghm_complete) +
plot_layout(widths = c(0.4,0.6))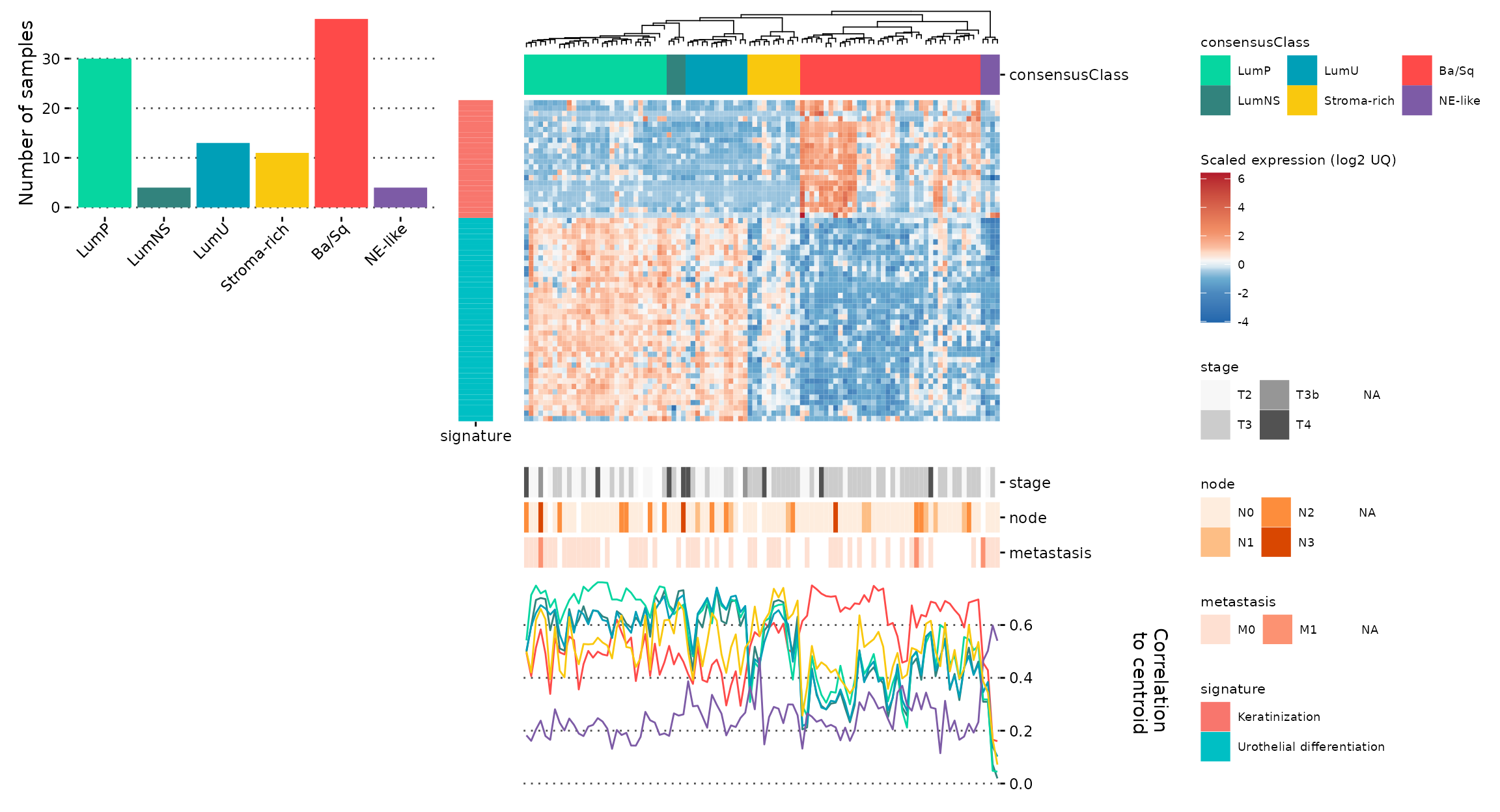
Row facetting
As of ggheatmapper 0.1.2, we’ve added row facetting options using the
rowv parameter. ggheatmapper will render parts of the
heatmap in different facets if the rowv argument for the
ggheatmap call is a named list:
sig_list <- split(tcgaBLCA_ex$gene_annot$gene_symbol, tcgaBLCA_ex$gene_annot$signature)
gr_gghm <- ggheatmap(tcgaBLCA_tb,
colv = "sample",
rowv = sig_list,
hm_colors = 'RdBu',
hm_color_values = scales::rescale(c(-4,-2,-1,-0.5,-0.25,0,0.25,0.5,1,2,4,6)),
scale = TRUE,
center = TRUE,
show_dend_row = FALSE,
show_colnames = FALSE,
show_rownames = FALSE,
group_colors = c(`Ba/Sq` = "#fe4a49", LumNS = "#32837d", LumP = "#06d6a0", LumU = "#009fb7",
`Stroma-rich` = "#f9c80e", `NE-like` = "#7d5ba6"),
colors_title = "Scaled expression (log2 UQ)")
gr_gghm +
plot_layout(guides = 'collect')